A study in boolean model parameterization
Last updated: 11 December, 2020
Intro
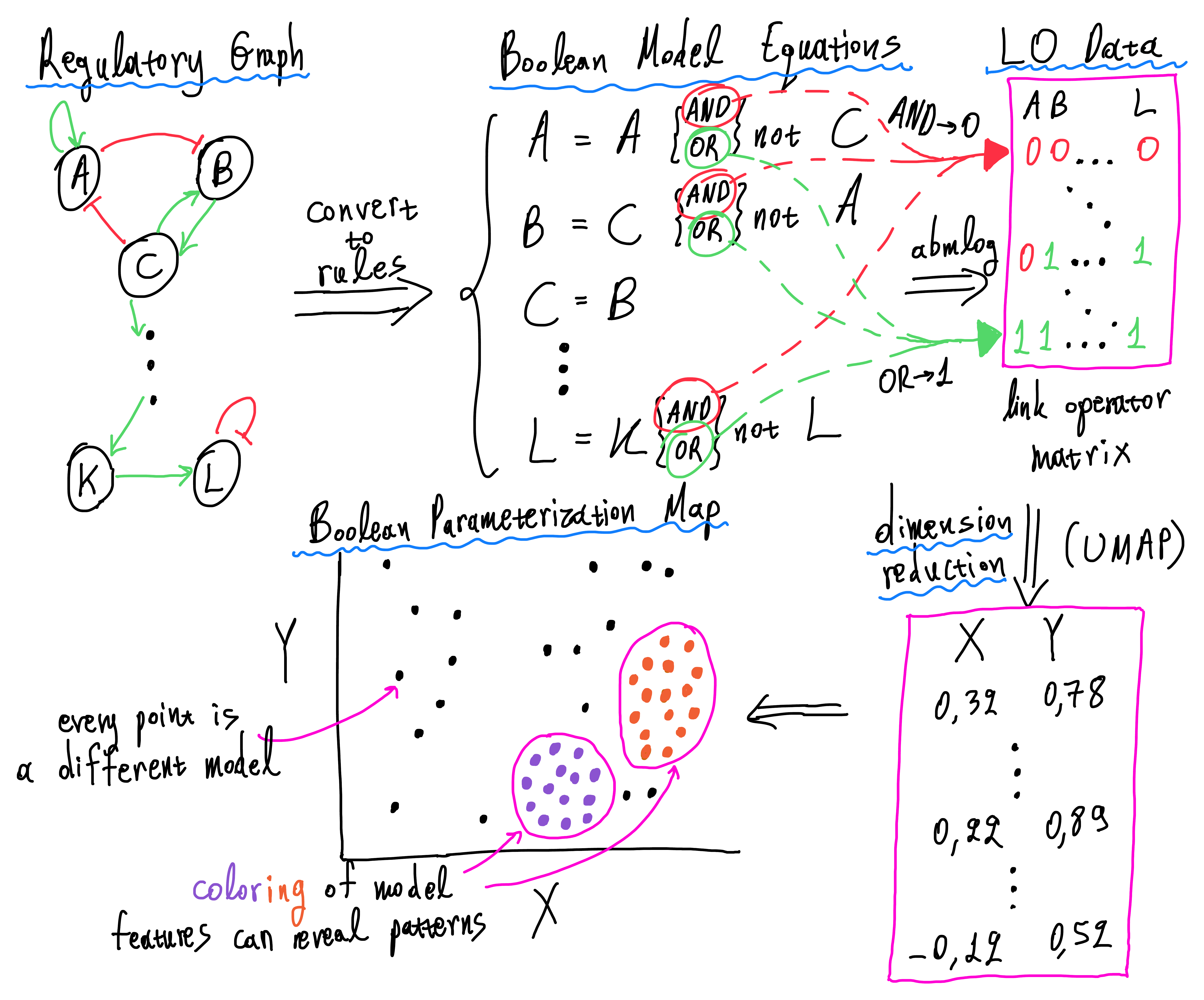
The purpose of this analysis is to make an investigation regarding the relation of boolean model parameterization (based on the standardized equation form inspired by (Mendoza and Xenarios 2006)) and various other model attributes, e.g. their drug combination prediction performance, their ability to predict synergies, their fitness to a specific cancerous cell line activity profile, as well as more general directives, such as the identification of essential nodes that drive the change of dynamics throughout the parameterization space.
The dataset used in this analysis is derived from the CASCADE 1.0 topology (Tsirvouli et al. 2020). The cancer signaling network has a total of \(77\) nodes, out of which \(23\) have link operators in their respective boolean equation (both activating and inhibiting regulators). Using the abmlog software, we generated all \(2^{23} = 8388608\) possible link operator mutated models for the CASCADE 1.0 topology.
We use the relative new, non-linear dimension reduction method UMAP (McInnes, Healy, and Melville 2018) to place all the generated boolean models in a boolean parameterization map and most of our conclusions are based on observing patterns in the produced maps.
The model dataset is available in Zenodo ![]() .
.
Libraries used in this analysis:
library(xfun)
library(knitr)
library(dplyr)
library(tidyr)
library(tibble)
library(ggpubr)
library(stringr)
library(ggplot2)
library(DT)
library(usefun)
library(emba)
library(forcats)
library(scales)
suppressPackageStartupMessages(library(ComplexHeatmap))
suppressPackageStartupMessages(library(circlize))
library(glmnet)
library(randomForest)
library(ranger)
library(uwot)
library(Ckmeans.1d.dp)